vignettes/hictkR-vignette.Rmd
hictkR-vignette.RmdOpening files
hictkR supports opening .hic and .cool files using the
File() function.
When opening files in .hic format, resolution is a
mandatory parameter.
path <- system.file("extdata", "interactions.hic", package = "hictkR")
f <- File(path, resolution = 100000)hictkR file handles returned by File() provide several
attributes storing static properties of the opened file
path <- f$path
resolution <- as.integer(f$resolution)
paste("File \"",
basename(path),
"\" has the following resolution: ",
resolution,
"bp",
sep = ""
)File “interactions.hic” has the following resolution: 100000 bp
Accessing chromosomes and bins
Some of these properties store the list of chromosomes as well as the bin table associated with the opened file.
chroms <- f$chromosomes
bins <- f$bins
normalizations <- f$normalizations| name | chr2L | chr2R | chr3L | chr3R | chr4 | chrX | chrY | chrM |
| size | 23513712 | 25286936 | 28110227 | 32079331 | 1348131 | 23542271 | 3667352 | 19524 |
| chrom | chr2L | chr2L | chr2L | chr2L | chr2L | chr2L | … |
| start | 0 | 1e+05 | 2e+05 | 3e+05 | 4e+05 | 5e+05 | … |
| end | 1e+05 | 2e+05 | 3e+05 | 4e+05 | 5e+05 | 6e+05 | … |
| ICE |
Fetching interactions
hictkR support fetching interactions through the fetch()
function.
Fetch genome-wide interactions with genomic coordinates
fetch(f, join = TRUE)| chrom1 | start1 | end1 | chrom2 | start2 | end2 | count |
|---|---|---|---|---|---|---|
| chr2L | 0 | 100000 | chr2L | 0 | 100000 | 101360 |
| chr2L | 0 | 100000 | chr2L | 100000 | 200000 | 29915 |
| chr2L | 0 | 100000 | chr2L | 200000 | 300000 | 4996 |
| chr2L | 0 | 100000 | chr2L | 300000 | 400000 | 2252 |
| chr2L | 0 | 100000 | chr2L | 400000 | 500000 | 3705 |
| chr2L | 0 | 100000 | chr2L | 500000 | 600000 | 2446 |
Interactions can also be normalized using one of the available
normalization methods (see f$normalizations).
fetch(f, join = TRUE, normalization = "ICE")| chrom1 | start1 | end1 | chrom2 | start2 | end2 | count |
|---|---|---|---|---|---|---|
| chr2L | 0 | 100000 | chr2L | 0 | 100000 | 0.6915117 |
| chr2L | 0 | 100000 | chr2L | 100000 | 200000 | 0.1748966 |
| chr2L | 0 | 100000 | chr2L | 200000 | 300000 | 0.0373811 |
| chr2L | 0 | 100000 | chr2L | 300000 | 400000 | 0.0238225 |
| chr2L | 0 | 100000 | chr2L | 400000 | 500000 | 0.0194293 |
| chr2L | 0 | 100000 | chr2L | 500000 | 600000 | 0.0158976 |
Fetch interactions for a region of interest
hictkR can be instructed to fetch interactions for a region of interest.
This is much more efficient than fetching genome-wide interactions and then filtering interactions using R.
fetch(f, "chr2L", join = TRUE)| chrom1 | start1 | end1 | chrom2 | start2 | end2 | count |
|---|---|---|---|---|---|---|
| chr2L | 0 | 100000 | chr2L | 0 | 100000 | 101360 |
| chr2L | 0 | 100000 | chr2L | 100000 | 200000 | 29915 |
| chr2L | 0 | 100000 | chr2L | 200000 | 300000 | 4996 |
| chr2L | 0 | 100000 | chr2L | 300000 | 400000 | 2252 |
| chr2L | 0 | 100000 | chr2L | 400000 | 500000 | 3705 |
| chr2L | 0 | 100000 | chr2L | 500000 | 600000 | 2446 |
fetch(f,
"chr2L:0-10,000,000",
"chr3R:10,000,000-20,000,000",
join = TRUE
)| chrom1 | start1 | end1 | chrom2 | start2 | end2 | count |
|---|---|---|---|---|---|---|
| chr2L | 0 | 100000 | chr3R | 10000000 | 10100000 | 55 |
| chr2L | 0 | 100000 | chr3R | 10100000 | 10200000 | 52 |
| chr2L | 0 | 100000 | chr3R | 10200000 | 10300000 | 26 |
| chr2L | 0 | 100000 | chr3R | 10300000 | 10400000 | 31 |
| chr2L | 0 | 100000 | chr3R | 10400000 | 10500000 | 22 |
| chr2L | 0 | 100000 | chr3R | 10500000 | 10600000 | 22 |
Fetch interactions as a dense matrix
hictkR can return interactions as a Matrix when
specifying type="dense".
fetch(f,
"chr2L:0-1,000,000",
"chr3R:1,000,000-2,000,000",
type = "dense"
)
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
#> [1,] 11 4 9 7 10 1 15 3 5 29
#> [2,] 11 10 15 4 14 7 5 2 12 44
#> [3,] 6 5 11 8 12 8 6 3 12 30
#> [4,] 6 2 8 3 5 7 1 2 7 15
#> [5,] 9 9 12 11 18 12 9 2 11 46
#> [6,] 17 7 13 4 16 8 1 4 9 64
#> [7,] 2 2 4 4 5 5 5 0 8 20
#> [8,] 3 6 11 4 11 7 3 1 9 19
#> [9,] 17 6 7 4 11 9 3 1 14 76
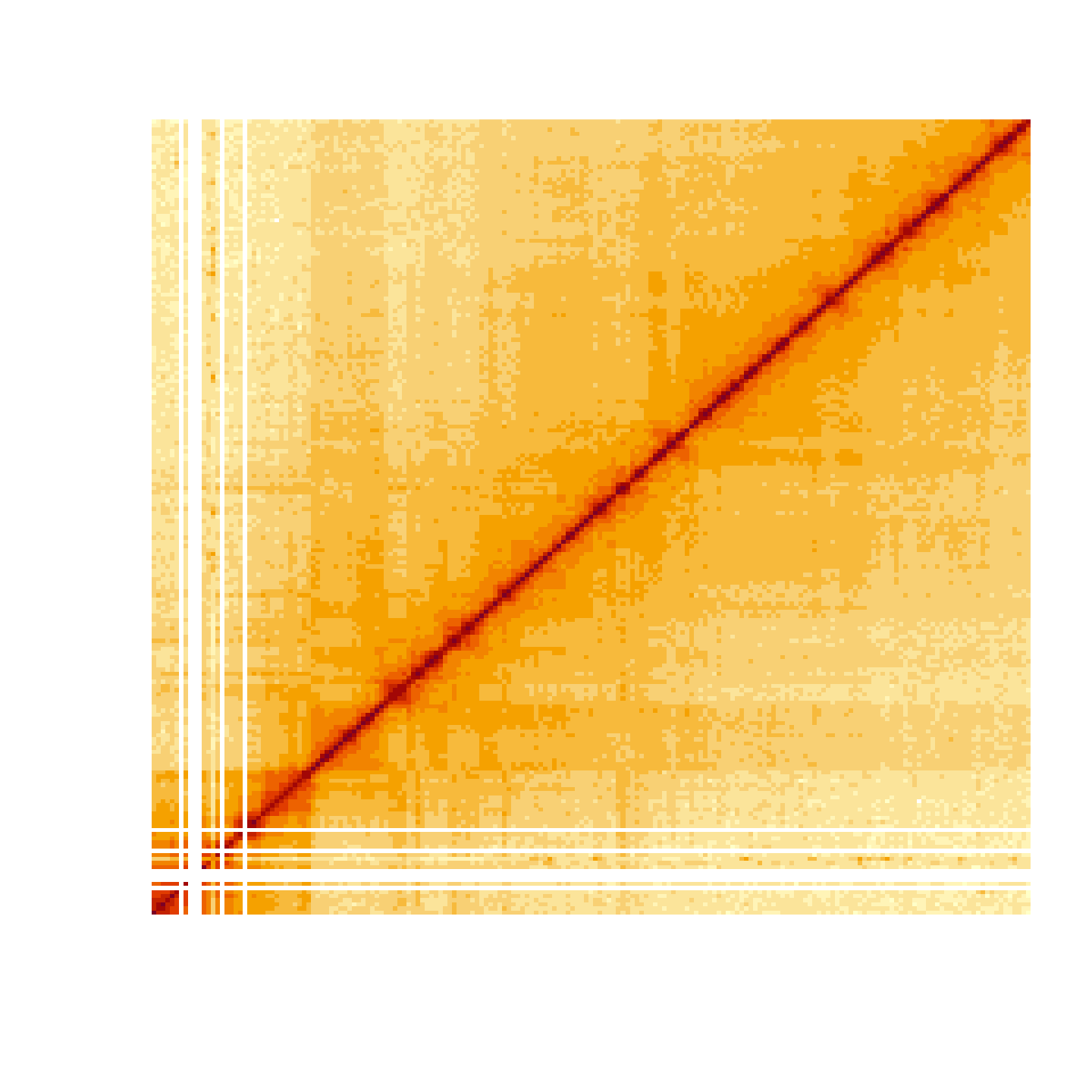
#> [10,] 0 2 9 2 7 2 4 1 3 7This can be very useful to visualize interaction matrices as heatmaps.
m <- fetch(f, "chr3R:0-20,000,000", normalization = "ICE", type = "dense")
image(log(m), axes = FALSE)